/ Nov 05, 2025
Trending
QUINN RIVET, ND
Dedicated to my father – Hugh Edmond Rivet
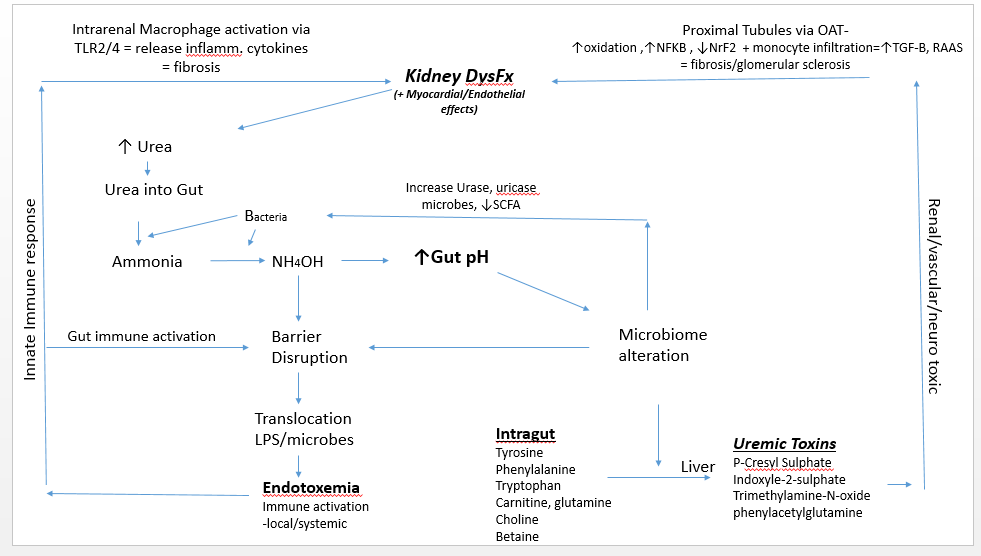
Evolving data over the last 40 years confirm that uremic metabolites generated by a dysbiotic microbiome impair kidney function and establish a bidirectional relationship between the gut and kidney. The altered microbiome and increased concentration of ammonium hydroxide (NH4OH) derived from transported urea into the gut causes a disruption of colonic transepithelial tight junctions, allowing toxicants and antigens to enter the systemic circulation.1,2 The result is endotoxemic inflammation, immune activation, and infiltration of the renal parenchyma, which have been related to the high cardiovascular morbidity of patients with kidney disease.
Immune activation ultimately suppresses innate and adaptive immunity, a process that corresponds with the concept of endotoxin tolerance, or immune paralysis. As in cases of advanced sepsis or chronic infection, this leaves the advanced renal patient with elevated urea open to infections.3,4 As the kidney is damaged, it loses functional capacity, further elevating urea levels and establishing a substantial bidirectional relationship between the kidney and the gut. This relationship is known as the gut-kidney axis.
This article will attempt to elucidate the kidney-gut axis, its role in progression or initiation of kidney disease, and the mechanisms at play, thus revealing reasonable therapeutic targets for the clinician.
Pathophysiology
A uremic gut milieu starts when a decline in renal function contributes to urea elevation in the systemic circulation. Urea then enters the gut via urea transporter-B.5 In the gut, urea is transformed into ammonia by bacteria, and then converted to ammonium hydroxide, which causes an increase in gut pH. In this environment, pathogenic microbes proliferate, particularly those that produce urease and uricase. This contributes to the eventual accumulation of various uremic toxins, including p-cresyl sulphate, indoxyl sulphate, and phenylacetylglutamine. In addition, the level of short-chain fatty acids (SCFAs) decreases. Taken together, these actions play a role in progressive renal fibrosis and glomerular sclerosis.6
In the late 1970s, Simenhoff et al and Vaziri et al revealed that people with chronic kidney disease (CKD) have markedly altered gut flora.6,7 Wong et al revealed that patients with end-stage renal disease (ESRD) harbor high gut populations of urease- and uricase-producing bacteria as well as bacteria that produce enzymes forming indole and p-cresol.8 These bacteria cause the fermentation of tyrosine and tryptophan, consequently increasing the circulatory concentration of indoxyl sulphate, p-cresyl, and p-cresyl sulphate.9 Once these uremic toxins reach the kidney’s proximal tubule, they are taken up by the organic anion transporter.10 The absorption of these toxins into tubules has several effects: the subsequent upregulation of nuclear factor-kappa beta (NF-kB),11 increased oxidation,12,13 decreased levels of the transcription factor NrF2,14 decreased mitochondrial function,15 upregulation of the renin-angiotensin system, and elaboration of transforming growth factor beta.16 All these actions are related to the progression of renal/tubulointerstitial fibrosis and a subsequent decline in kidney function. These urotoxins also lead to hypermethylation and inhibition of Klotho gene expression. (The Klotho gene is required for protection of the kidney; its inhibition is linked to the acceleration of renal function decline.)17
In this gut milieu, choline, L-carnitine, and betaine are metabolized to trimethylamine, which is then metabolized in the liver by flavin-containing monooxygenases. This process produces trimethylamine N-oxide, a uremic toxin associated with atherosclerosis and increased mortality rates in ESRD patients.18 Phenylacetylglutamine, another uremic toxin family, has been related to poor patient survival with those who have CKD19,20 as well as stroke.21
Another factor in the uremic gut scenario is decreased SCFA levels. SCFAs regulate kidney function via activation of trans-membrane G protein-coupled receptors and inhibition of histone acetylation.22 SCFAs modulate inflammation, blood pressure, oxidative stress, immune balance, and glucose metabolism – all of which are undermined by the progressive loss of renal function.22,23
Elevated concentrations of NH4OH in the gut leads to a loss of integrity of the gut barrier.5 This disruption causes local neutrophilic infiltration and allows for the translocation of gut-derived antigens such as lipopolysaccharides and microbes. This can result in overt endotoxemia, with subsequent innate immune activation and persistent systemic inflammation. Potential consequences include atherosclerosis, cardiomyocyte aberrations contributing to arrhythmias, intracardial fibrosis, protein wasting, and increased morbidity and mortality.5, 24-25 Translocated antigens can coordinate with toll-like receptors-2 or 4 (TLRs) on renal macrophages, which then express inflammatory cytokines and chemokines to produce an inflammatory cascade in the kidney.25,26 TLRs are key regulators of the innate immune system; their activation is related to oxidative stress, renal injury secondary to activation of the renin-angiotensin system, hypertension, and cardiovascular disease.27
In summary, as renal function declines, urea enters the gut via the circulation and creates a milieu favorable for pathogenic bacteria. Uremic toxins from these bacteria increase and SCFA levels decrease. This allows for renal parenchyma fibrosis and vasculature dysfunction/sclerosis, which activates intrarenal immune cells. Intrarenal inflammation and dysfunction result, causing associated uremia. This process illustrates the cyclical axis established between the kidney and gut.
Disease Entities
Certain kidney disease entities – namely IgA nephropathy (IgAN),28-42 focal segmental glomerulosclerosis (FSGS),42-44 and lupus nephritis (LN)45-47 – may be the result of a primary gut microbial disturbance with localized genetic predispositions. Environmental triggers may lead to these conditions in those with a genetic predisposition or dysbiotic gut microbiome. Progressive kidney dysfunction allows for the elevation of urea and the production of gut-derived uremic toxins, the effects of which are relative to the decline in renal function and associated progression of fibrosis.
IgAN, which has been linked to certain genetic traits, results in the elaboration of abnormal mucosal IgA.48 In IgAN, IgA lacks a galactose molecule and is O-glycosylated,49 which renders it antigenic; when put into circulation, this altered IgA can thus result in the formation of immune complexes that deposit in the renal mesangium. By triggering the complement cascade, these complexes initiate intrarenal immune activation with resultant inflammation and the characteristic clinical entities of proteinuria and hematuria.49 In genetically prone individuals, various mucosal immune activators can cause robust production of the galactose-deficient IgA. Examples include wheat proteins, casein, soya, and mucosal infections.50-56
Certain patient populations that have been diagnosed with FSGS may have a gut/immune-derived circulating factor known as soluble urokinase plasminogen activating receptor.57 This receptor binds to a cellular anchoring molecule (alpha 3 beta 1 integrin) in the electron-dense membrane junction of the podocyte (part of the filtration mechanism in the glomerulus) that is galactose based. The result is effacement of the podocyte and subsequent nephrotic-range proteinuria.58 Hypoallergenic and oral galactose has been used in patients refractory to steroid therapy as well as those suffering recurrent FSGS post-transplantation.59
In LN, a perturbed gut microbiota has been reported involving the phenomenon of molecular mimicry. Orthologs of human Ro60 autoantigen have been observed in gut bacteria in patients with systemic lupus erythematosus (SLE).60 If translocated, this would then result in a T-cell reaction and production of anti-Ro60 autoantibodies. Specifically, Ruminococcus gnavus, found in the guts of many SLE patients, cross-reacts with human DNA and is positively correlated with SLE and thus LN.47,60
Related Studies
The inculcation of gut-focused therapeutics for the nephrology patient has yet to occur in nephrology clinics in North America, even though published scientific evidence clearly shows a pathophysiological relationship between the gut and kidney.
Zheng et al61 reviewed studies from January 1, 2000, to May 15, 2019, which included 13 trials and a combined total of 671 patients. This meta-analysis supported the use of probiotics, prebiotics, and synbiotics (a combination of both), showing improvement of biomarkers of inflammation and oxidative stress as well as lipid profiles. The duration of intervention, BMI, type of intervention, and age influenced the outcome of microbial therapies. Bakhtiary et al62 reviewed studies through April 2020 and 14 randomized controlled trials were included. In patients with CKD, pre/pro/synbiotics significantly reduced total cholesterol, fasting blood glucose, insulin resistance, insulin levels, high-sensitivity C-reactive protein (hs-CRP), and malondialdehyde compared to controls. Interventions increased insulin sensitivity, total antioxidant capacity, and glutathione levels. Bohlouli et al63 reviewed the effects of probiotics on oxidative stress and inflammatory status in patients with diabetic nephropathy; this review included 7 trials and 340 total participants. Findings revealed that supplementation with probiotics decreased CRP and malondialdehyde, while glutathione and total antioxidant capacity were increased. McFarlane et al64 reviewed 16 studies through 2017 investigating 645 adults. Of these, 5 investigated prebiotics, 6 investigated probiotics, and 5 investigated synbiotics. The results showed that these interventions largely made little to no difference in levels of serum urea, indoxyl sulphate, and p-cresyl sulphate. However, in 4 studies involving 105 participants, prebiotics possibly decreased serum urea. The review did not investigate oxidative or inflammatory markers.64
Vlachou et al65 did a systematic review of studies using probiotics on diabetic nephropathy patients. This review found that most studies demonstrated benefit, including reduction of inflammation, oxidative stress, and renal function biomarkers. Main microorganisms used belonged to Lactobacillus and Bifidobacterium genera. Dosages ranged from 2×10^^7 to 6×10^^10 CFU/g. Fagundes et al66 looked at studies examining the effects of probiotics on CKD, conducted between 2012 and 2016 in English and Portuguese, and found most studies reporting positive outcomes in renal function, including a decrease in blood urea nitrogen (BUN), ammonia, p-cresol, p-cresyl sulphate, and indoxyl-sulphate. In a 2018 systematic review of probiotics by Jia et al,67 8 studies were evaluated that included 261 total patients in CKD stages 3-5. A decrease in p-cresyl sulphate was found in 125 patients (3/8 of the studies) and an increase in interleukin-6 (IL-6) was found in 134 patients (also 3/8 studies). No significant differences were found in BUN or creatinine. McFarlane, C. et al64 analyzed studies using pro/pre/synbiotics from 2000 to 2017 and found no significant differences in terms of serum urea or indoxyl or p-cresyl sulphate. Prebiotics did, however, produce a slight decrease in urea. Clinical effects were deemed “uncertain.”
In a study by Lai et al68, a small intervention group (18/41) received a low-protein diet (0.6 g/kg) plus 19 g/d of inulin for 6 months, while a control group only received a low-protein diet. At 6 months, both groups showed a significantly lower serum urea and phosphorus, uric acid, and metabolic acidosis. However, the intervention group also showed a reduction in serum insulin, fasting glucose, total serum cholesterol, triglycerides, homocysteine, and CRP, and an increase in HDL. The intervention group also reported an improvement in mood. Melekoglu et al69 performed a study on rats with induced CKD (n = 16). Supplemented with an inulin-based prebiotic at a dose of 5g/kg/day, these rats demonstrated reduced p-cresyl, urea, and IL-6, but not indoxyl sulphate. They also demonstrated enhanced antioxidant capacity, but the prebiotic did not show any observable effect on the colonic epithelial tight-junction proteins, claudin-1 and occludin. Zirker70 did a good overview of prebiotics, their effects, and some food products that contain them.
AST-120 is an oral spherical carbon adsorbent consisting of porous particles that are 0.2-0.4 mm in diameter, insoluble in water, and able to absorb both acidic and basic compounds.71 Su P. et al72 reviewed 15 studies of AST-120 supplementation. With a patient aggregate of 3763, the meta-analysis revealed “non-significant differences in mortality” among all treatment groups, including those receiving no AST-120. However, when dosages were “tailored” (3.0–7.2 g/d), event rates of ESRD and composite renal outcomes were significantly lower. Asai et al71 looked at clinical data related to AST-120, from 1982 to 2013. The results suggest that AST-120 may delay both kidney function decline and the initiation of dialysis.
Clinical Intervention
Overall, the studies evaluated that used just probiotics showed lower inflammation and increased antioxidant capacity but no significant differences in uremic toxins. Studies that included prebiotics/fiber showed a decrease in uremic toxins and inflammatory markers with significant lowering of lipids and increase in mentation. From the published evidence, an optimal clinical intervention would include:
As with most naturopathic approaches, based on the relevant scientific literature, the synergy between all applications given will most likely produce the best results for the kidney patient. Until the utility of gut-focused therapeutics can become more widely adopted, it may be up to the naturopathic practitioner to shore up this deficiency within the nephrology clinic.
References

Quinn Rivet, ND graduated from CCNM in 1994. From 2001 to 2012 he was an instructor, clinical supervisor, and the chair of Nutrition (2004-2010). After returning from 3 years in China, he now pursues further work in clinical research, education, and consultation services. Dr. Rivet can be contacted through his web site: drquinn-nd.com.
QUINN RIVET, ND
Dedicated to my father – Hugh Edmond Rivet
Evolving data over the last 40 years confirm that uremic metabolites generated by a dysbiotic microbiome impair kidney function and establish a bidirectional relationship between the gut and kidney. The altered microbiome and increased concentration of ammonium hydroxide (NH4OH) derived from transported urea into the gut causes a disruption of colonic transepithelial tight junctions, allowing toxicants and antigens to enter the systemic circulation.1,2 The result is endotoxemic inflammation, immune activation, and infiltration of the renal parenchyma, which have been related to the high cardiovascular morbidity of patients with kidney disease.
Immune activation ultimately suppresses innate and adaptive immunity, a process that corresponds with the concept of endotoxin tolerance, or immune paralysis. As in cases of advanced sepsis or chronic infection, this leaves the advanced renal patient with elevated urea open to infections.3,4 As the kidney is damaged, it loses functional capacity, further elevating urea levels and establishing a substantial bidirectional relationship between the kidney and the gut. This relationship is known as the gut-kidney axis.
This article will attempt to elucidate the kidney-gut axis, its role in progression or initiation of kidney disease, and the mechanisms at play, thus revealing reasonable therapeutic targets for the clinician.
Pathophysiology
A uremic gut milieu starts when a decline in renal function contributes to urea elevation in the systemic circulation. Urea then enters the gut via urea transporter-B.5 In the gut, urea is transformed into ammonia by bacteria, and then converted to ammonium hydroxide, which causes an increase in gut pH. In this environment, pathogenic microbes proliferate, particularly those that produce urease and uricase. This contributes to the eventual accumulation of various uremic toxins, including p-cresyl sulphate, indoxyl sulphate, and phenylacetylglutamine. In addition, the level of short-chain fatty acids (SCFAs) decreases. Taken together, these actions play a role in progressive renal fibrosis and glomerular sclerosis.6
In the late 1970s, Simenhoff et al and Vaziri et al revealed that people with chronic kidney disease (CKD) have markedly altered gut flora.6,7 Wong et al revealed that patients with end-stage renal disease (ESRD) harbor high gut populations of urease- and uricase-producing bacteria as well as bacteria that produce enzymes forming indole and p-cresol.8 These bacteria cause the fermentation of tyrosine and tryptophan, consequently increasing the circulatory concentration of indoxyl sulphate, p-cresyl, and p-cresyl sulphate.9 Once these uremic toxins reach the kidney’s proximal tubule, they are taken up by the organic anion transporter.10 The absorption of these toxins into tubules has several effects: the subsequent upregulation of nuclear factor-kappa beta (NF-kB),11 increased oxidation,12,13 decreased levels of the transcription factor NrF2,14 decreased mitochondrial function,15 upregulation of the renin-angiotensin system, and elaboration of transforming growth factor beta.16 All these actions are related to the progression of renal/tubulointerstitial fibrosis and a subsequent decline in kidney function. These urotoxins also lead to hypermethylation and inhibition of Klotho gene expression. (The Klotho gene is required for protection of the kidney; its inhibition is linked to the acceleration of renal function decline.)17
In this gut milieu, choline, L-carnitine, and betaine are metabolized to trimethylamine, which is then metabolized in the liver by flavin-containing monooxygenases. This process produces trimethylamine N-oxide, a uremic toxin associated with atherosclerosis and increased mortality rates in ESRD patients.18 Phenylacetylglutamine, another uremic toxin family, has been related to poor patient survival with those who have CKD19,20 as well as stroke.21
Another factor in the uremic gut scenario is decreased SCFA levels. SCFAs regulate kidney function via activation of trans-membrane G protein-coupled receptors and inhibition of histone acetylation.22 SCFAs modulate inflammation, blood pressure, oxidative stress, immune balance, and glucose metabolism – all of which are undermined by the progressive loss of renal function.22,23
Elevated concentrations of NH4OH in the gut leads to a loss of integrity of the gut barrier.5 This disruption causes local neutrophilic infiltration and allows for the translocation of gut-derived antigens such as lipopolysaccharides and microbes. This can result in overt endotoxemia, with subsequent innate immune activation and persistent systemic inflammation. Potential consequences include atherosclerosis, cardiomyocyte aberrations contributing to arrhythmias, intracardial fibrosis, protein wasting, and increased morbidity and mortality.5, 24-25 Translocated antigens can coordinate with toll-like receptors-2 or 4 (TLRs) on renal macrophages, which then express inflammatory cytokines and chemokines to produce an inflammatory cascade in the kidney.25,26 TLRs are key regulators of the innate immune system; their activation is related to oxidative stress, renal injury secondary to activation of the renin-angiotensin system, hypertension, and cardiovascular disease.27
In summary, as renal function declines, urea enters the gut via the circulation and creates a milieu favorable for pathogenic bacteria. Uremic toxins from these bacteria increase and SCFA levels decrease. This allows for renal parenchyma fibrosis and vasculature dysfunction/sclerosis, which activates intrarenal immune cells. Intrarenal inflammation and dysfunction result, causing associated uremia. This process illustrates the cyclical axis established between the kidney and gut.
Disease Entities
Certain kidney disease entities – namely IgA nephropathy (IgAN),28-42 focal segmental glomerulosclerosis (FSGS),42-44 and lupus nephritis (LN)45-47 – may be the result of a primary gut microbial disturbance with localized genetic predispositions. Environmental triggers may lead to these conditions in those with a genetic predisposition or dysbiotic gut microbiome. Progressive kidney dysfunction allows for the elevation of urea and the production of gut-derived uremic toxins, the effects of which are relative to the decline in renal function and associated progression of fibrosis.
IgAN, which has been linked to certain genetic traits, results in the elaboration of abnormal mucosal IgA.48 In IgAN, IgA lacks a galactose molecule and is O-glycosylated,49 which renders it antigenic; when put into circulation, this altered IgA can thus result in the formation of immune complexes that deposit in the renal mesangium. By triggering the complement cascade, these complexes initiate intrarenal immune activation with resultant inflammation and the characteristic clinical entities of proteinuria and hematuria.49 In genetically prone individuals, various mucosal immune activators can cause robust production of the galactose-deficient IgA. Examples include wheat proteins, casein, soya, and mucosal infections.50-56
Certain patient populations that have been diagnosed with FSGS may have a gut/immune-derived circulating factor known as soluble urokinase plasminogen activating receptor.57 This receptor binds to a cellular anchoring molecule (alpha 3 beta 1 integrin) in the electron-dense membrane junction of the podocyte (part of the filtration mechanism in the glomerulus) that is galactose based. The result is effacement of the podocyte and subsequent nephrotic-range proteinuria.58 Hypoallergenic and oral galactose has been used in patients refractory to steroid therapy as well as those suffering recurrent FSGS post-transplantation.59
In LN, a perturbed gut microbiota has been reported involving the phenomenon of molecular mimicry. Orthologs of human Ro60 autoantigen have been observed in gut bacteria in patients with systemic lupus erythematosus (SLE).60 If translocated, this would then result in a T-cell reaction and production of anti-Ro60 autoantibodies. Specifically, Ruminococcus gnavus, found in the guts of many SLE patients, cross-reacts with human DNA and is positively correlated with SLE and thus LN.47,60
Related Studies
The inculcation of gut-focused therapeutics for the nephrology patient has yet to occur in nephrology clinics in North America, even though published scientific evidence clearly shows a pathophysiological relationship between the gut and kidney.
Zheng et al61 reviewed studies from January 1, 2000, to May 15, 2019, which included 13 trials and a combined total of 671 patients. This meta-analysis supported the use of probiotics, prebiotics, and synbiotics (a combination of both), showing improvement of biomarkers of inflammation and oxidative stress as well as lipid profiles. The duration of intervention, BMI, type of intervention, and age influenced the outcome of microbial therapies. Bakhtiary et al62 reviewed studies through April 2020 and 14 randomized controlled trials were included. In patients with CKD, pre/pro/synbiotics significantly reduced total cholesterol, fasting blood glucose, insulin resistance, insulin levels, high-sensitivity C-reactive protein (hs-CRP), and malondialdehyde compared to controls. Interventions increased insulin sensitivity, total antioxidant capacity, and glutathione levels. Bohlouli et al63 reviewed the effects of probiotics on oxidative stress and inflammatory status in patients with diabetic nephropathy; this review included 7 trials and 340 total participants. Findings revealed that supplementation with probiotics decreased CRP and malondialdehyde, while glutathione and total antioxidant capacity were increased. McFarlane et al64 reviewed 16 studies through 2017 investigating 645 adults. Of these, 5 investigated prebiotics, 6 investigated probiotics, and 5 investigated synbiotics. The results showed that these interventions largely made little to no difference in levels of serum urea, indoxyl sulphate, and p-cresyl sulphate. However, in 4 studies involving 105 participants, prebiotics possibly decreased serum urea. The review did not investigate oxidative or inflammatory markers.64
Vlachou et al65 did a systematic review of studies using probiotics on diabetic nephropathy patients. This review found that most studies demonstrated benefit, including reduction of inflammation, oxidative stress, and renal function biomarkers. Main microorganisms used belonged to Lactobacillus and Bifidobacterium genera. Dosages ranged from 2×10^^7 to 6×10^^10 CFU/g. Fagundes et al66 looked at studies examining the effects of probiotics on CKD, conducted between 2012 and 2016 in English and Portuguese, and found most studies reporting positive outcomes in renal function, including a decrease in blood urea nitrogen (BUN), ammonia, p-cresol, p-cresyl sulphate, and indoxyl-sulphate. In a 2018 systematic review of probiotics by Jia et al,67 8 studies were evaluated that included 261 total patients in CKD stages 3-5. A decrease in p-cresyl sulphate was found in 125 patients (3/8 of the studies) and an increase in interleukin-6 (IL-6) was found in 134 patients (also 3/8 studies). No significant differences were found in BUN or creatinine. McFarlane, C. et al64 analyzed studies using pro/pre/synbiotics from 2000 to 2017 and found no significant differences in terms of serum urea or indoxyl or p-cresyl sulphate. Prebiotics did, however, produce a slight decrease in urea. Clinical effects were deemed “uncertain.”
In a study by Lai et al68, a small intervention group (18/41) received a low-protein diet (0.6 g/kg) plus 19 g/d of inulin for 6 months, while a control group only received a low-protein diet. At 6 months, both groups showed a significantly lower serum urea and phosphorus, uric acid, and metabolic acidosis. However, the intervention group also showed a reduction in serum insulin, fasting glucose, total serum cholesterol, triglycerides, homocysteine, and CRP, and an increase in HDL. The intervention group also reported an improvement in mood. Melekoglu et al69 performed a study on rats with induced CKD (n = 16). Supplemented with an inulin-based prebiotic at a dose of 5g/kg/day, these rats demonstrated reduced p-cresyl, urea, and IL-6, but not indoxyl sulphate. They also demonstrated enhanced antioxidant capacity, but the prebiotic did not show any observable effect on the colonic epithelial tight-junction proteins, claudin-1 and occludin. Zirker70 did a good overview of prebiotics, their effects, and some food products that contain them.
AST-120 is an oral spherical carbon adsorbent consisting of porous particles that are 0.2-0.4 mm in diameter, insoluble in water, and able to absorb both acidic and basic compounds.71 Su P. et al72 reviewed 15 studies of AST-120 supplementation. With a patient aggregate of 3763, the meta-analysis revealed “non-significant differences in mortality” among all treatment groups, including those receiving no AST-120. However, when dosages were “tailored” (3.0–7.2 g/d), event rates of ESRD and composite renal outcomes were significantly lower. Asai et al71 looked at clinical data related to AST-120, from 1982 to 2013. The results suggest that AST-120 may delay both kidney function decline and the initiation of dialysis.
Clinical Intervention
Overall, the studies evaluated that used just probiotics showed lower inflammation and increased antioxidant capacity but no significant differences in uremic toxins. Studies that included prebiotics/fiber showed a decrease in uremic toxins and inflammatory markers with significant lowering of lipids and increase in mentation. From the published evidence, an optimal clinical intervention would include:
As with most naturopathic approaches, based on the relevant scientific literature, the synergy between all applications given will most likely produce the best results for the kidney patient. Until the utility of gut-focused therapeutics can become more widely adopted, it may be up to the naturopathic practitioner to shore up this deficiency within the nephrology clinic.
References
Quinn Rivet, ND graduated from CCNM in 1994. From 2001 to 2012 he was an instructor, clinical supervisor, and the chair of Nutrition (2004-2010). After returning from 3 years in China, he now pursues further work in clinical research, education, and consultation services. Dr. Rivet can be contacted through his web site: drquinn-nd.com.